
 From the point of view of ab initio (first principles) electronic structure
methods, a basis set is simply a collection of functions, whose members are
typically associated with one or more of the atoms in a molecule. When people
say that they are "using the 3-21G basis on ethylene" they really mean that
they're performing a calculation with the appropriate carbon and hydrogen 3-21G
basis functions ("3-21G" is the just the name given to this basis set family
by scientists who originally developed it) positioned at the two carbons and
four hydrogens in C2H4, for a total of 26 functions.
From the point of view of ab initio (first principles) electronic structure
methods, a basis set is simply a collection of functions, whose members are
typically associated with one or more of the atoms in a molecule. When people
say that they are "using the 3-21G basis on ethylene" they really mean that
they're performing a calculation with the appropriate carbon and hydrogen 3-21G
basis functions ("3-21G" is the just the name given to this basis set family
by scientists who originally developed it) positioned at the two carbons and
four hydrogens in C2H4, for a total of 26 functions.
Basis sets are a mathematical convenience because the quantum mechanical equations which describe the behavior of electrons in molecules are most easily solved by expanding the wavefunction or density in terms of a finite set. Only in specialized cases, such as diatomic molecules, has it proven computationally feasible to forego the use of basis sets in favor of fully numerical techniques.
 Along with the sophistication of the approach used in describing the correlated
motions of the electrons in a molecule, basis sets represent one of the two
primary user-selectable input parameters for ab initio programs such
as Gaussian, GAMESS and NWChem. A poorly chosen basis set will typically lead
to large inaccuracies in the computed results or, in some cases, qualitatively
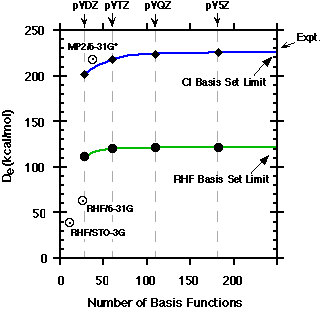
incorrect findings. A simple example is the dissociation energy of N2. The experimental
value of De is 228 kcal/mol, whereas small basis set RHF predicts a value of
39 kcal/mol. Larger basis sets, used with highly correlated methods can come
within 1 - 2 kcal/mol of experiment. Another example is the hydronium cation,
H3O+, which has a pyramidal shape like ammonia. With small basis sets this molecule
is incorrectly predicted to be flat.
Along with the sophistication of the approach used in describing the correlated
motions of the electrons in a molecule, basis sets represent one of the two
primary user-selectable input parameters for ab initio programs such
as Gaussian, GAMESS and NWChem. A poorly chosen basis set will typically lead
to large inaccuracies in the computed results or, in some cases, qualitatively
incorrect findings. A simple example is the dissociation energy of N2. The experimental
value of De is 228 kcal/mol, whereas small basis set RHF predicts a value of
39 kcal/mol. Larger basis sets, used with highly correlated methods can come
within 1 - 2 kcal/mol of experiment. Another example is the hydronium cation,
H3O+, which has a pyramidal shape like ammonia. With small basis sets this molecule
is incorrectly predicted to be flat.
Some basis sets consist of relatively few functions. For example, the STO-3G basis has only one function per occupied atomic orbital (1s, 2s, 2px, 2py, 2pz). Others have a large number of functions of different symmetries (e.g. s, p, d, f...). Basis sets which are too small may lack the flexibility to describe the basic physics of a problem and can produce qualitatively misleading results, with no hint of trouble. Likewise, overly large basis sets may waste many hours of computer time.
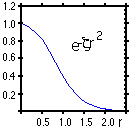
g(r) = N*(x l)*(y m)*(zeta n)*exp(-zeta*r 2),
where N is a normalization constant which insures that the square of the Gaussian gives a value of 1.0 when integrated over all space, (l,m,n) are integer powers of the electron's Cartesian coordinates ranging from 0 to some small positive value, and zeta is an exponent which helps determine the radial size of the function. The variable r represents the distance of the electron from the origin of the Gaussian.
Functions with L = l+m+n = 0 are spherically symmetric about the origin and are known as "s" functions. Similary, the three functions corresponding to l+m+n = 1 are the p(x), p(y), p(z) functions, etc. The Cartesian Gaussians possess six functions with l+m+n = 2, from which the five spherical components, d(xy), d(xz), d(yz), d(xx-yy) and d(2zz-xx-yy), can be constructed. The remaining function is of spherical symmetry and is customarily deleted. As the total L value increases, the difference in the number of Cartesian and spherical components increases. Many electronic structure programs are able to handle either form.
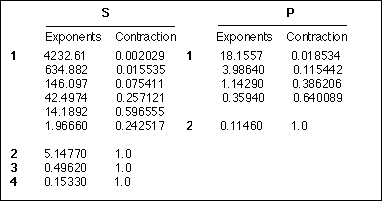
There are two types of contracted basis functions in widespread use today. In "segemented" contractions a given primitive function appears in no more than one contracted function. Two examples of segmented contractions are the STO-3G and the Dunning DZ basis sets, the latter of which is shown here for the carbon atom.
Segmented contractions became more difficult to construct as you went further down the periodic table. This led to the development of another way of contracting Gaussian primitives.
The second category of contracted basis function is referred to as a "general" contraction. In general contractions a given primitive can appear in more than one contracted function. Examples of generally contracted basis sets are the NASA Ames Atomic Natural Orbital (ANO) and the Roos ANO basis sets. An example from the latter family is shown to the right. Generally contracted basis sets have a reputation for being more time consuming to use than their segmented counterparts. This is partially due to the relative scarcity of integral programs which are designed to handle these basis functions efficiently. Although it is possible to use general contractions with any integral program, there will be a penalty to pay with some codes. For example, if there are 10 generally contracted s-symmetry basis functions defined in terms of 20 Gaussian primitives, some codes will interpret this situation as a calculation over 10 x 20 = 200 primitives.
Finally, some basis sets possess characteristics of both contraction styles. For the sake of simplicity, these sets are normally referred to as generally contracted basis sets. The new correlation consistent basis set family developed by T. H. Dunning, Jr. and co-workers is an example of a hybrid contraction style.
With this last category of contraction it is sometimes possible to reformat the basis set so that the performance penalty you're forced to pay for using generally contracted functions in programs that weren't designed for them is minimized. In the Ecce Basis Set Tool we refer to this reformatting as "optimizing" the general contraction.
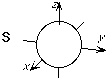
The general shapes of the spherical harmonic forms of the basis functions up through l = 3 (f functions) is shown here.
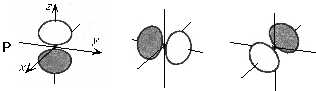
From a practical perspective, the first polarization functions (e.g. a set of d functions on carbon) are the most important additions one can make to the basis set beyond the valence s and p functions. At the Hartree-Fock level of theory, most properties converge to the complete basis set limit relatively quickly with the addition of more polarization functions. However, at the correlated level of theory the convergence is typically much slower, so that many higher l functions are needed in order to reach the complete basis set limit. In particularly difficult cases, such as the dissociation energy of N2, basis sets containing d and f polarization functions still underestimate the true value of De by more than 10 kcal/mol.
As is evident from these pictures, as the l value increases, so does the number of angular nodes (places where the orbital changes sign).
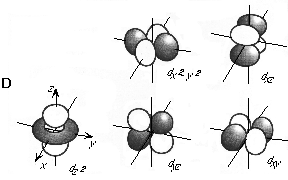
The l = 4 (g) functions have 4 such nodal planes. Although g functions are not shown, you can imagine that their 3D shapes have gotten to be quite complex and there are a lot of them. Because of the large number of g functions and the fact that integrals over g functions are time consuming to compute, relatively few polyatomic calculations are performed with these functions.
Some programs can perform calculations with two different forms of the
higher l value Gaussians. There are l*(l+1)/2 + (l+1) Cartesian Gaussians,
but only 2*l+1 Spherical Harmonic functions for a given value of l. Thus,
for l greater than 1 (p functions) there are more Cartesian than Spherical
components. The relative numbers of Cartesian and Spherical Gaussians are:
d functions (l=2) 6 Cart. and 5 Spher.,
f functions (l=3) 10 Cart. and 7 Spher.,
g functions (l=4) 15 Cart. and 9 Spher.,
h functions (l=5) 21 Cart. and 11 Spher.

A wide range of special polarization sets have been designed for various properties or types of calculations.
| Ecce Online Help Revised: November 3, 2002 |
Disclaimer |